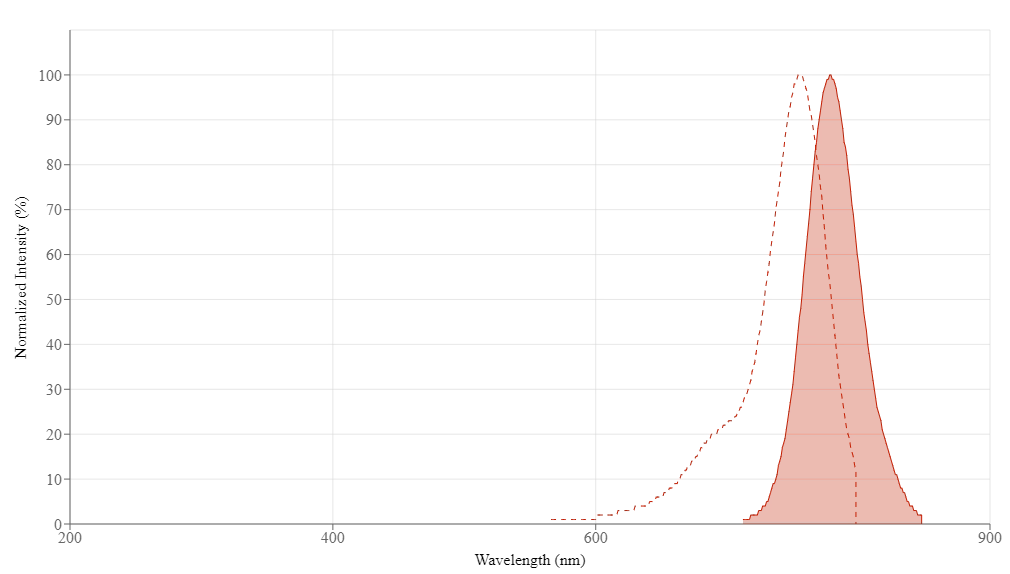
Fluorescent labelling with Cy7 has emerged as a cornerstone technology in advanced biomedical imaging, enabling researchers to visualize biological processes with unprecedented depth and clarity. As a member of the heptamethine cyanine dye family, Cy7 is characterized by its exceptional near-infrared (NIR) fluorescence properties, with excitation and emission maxima at approximately 749 nm and 776 nm, respectively. This spectral positioning within the NIR optical window (650–900 nm) minimizes interference from endogenous biomolecules like hemoglobin and water, allowing for deep tissue penetration of up to 15 cm and significantly reducing background autofluorescence. The strategic application of Cy7 labelling has revolutionized fields ranging from in vivo imaging and photodynamic therapy to drug delivery systems, with recent advances even witnessing a Cy7-based theranostic agent enter clinical trials. Understanding the principles, methodologies, and applications of Cy7 conjugation is therefore essential for researchers seeking to leverage this powerful tool in their investigations.
Key Takeaways
- Cy7 exhibits excitation/emission maxima at ~749/776 nm, placing it within the ideal NIR window for deep-tissue imaging with minimal background interference.
- The dye’s heptamethine structure enables dual functionality as both a fluorescent probe and a photosensitizer for photodynamic and photothermal therapies.
- Conjugation typically employs NHS ester chemistry targeting primary amines or maleimide chemistry for thiol-specific labelling, with reaction conditions carefully optimized to preserve biomolecular activity.
- Cy7-labelled peptides and proteins are indispensable tools in in vivo imaging, theranostic agent development, and studies of cell-cell interactions such as the LIPSTIC technique.
Fundamentals of Cy7 Structure and Photophysics
Chemical Architecture of Heptamethine Cyanine Dyes
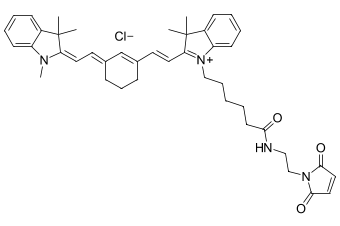
The molecular structure of Cy7 is defined by a central conjugated polymethine chain connecting two nitrogen-containing indole heterocycles. This heptamethine framework creates an extensive π-conjugated system responsible for the dye’s strong absorption in the NIR region. One indole moiety carries a positive charge, resulting in a delocalized cationic structure that influences both the dye’s photophysical behavior and its interaction with biological environments. This unique architecture not only confers exceptional brightness but also enables structural modifiability at multiple sites, allowing researchers to fine-tune properties such as water solubility, targeting specificity, and photosensitizing efficiency.
Spectral Advantages and the NIR Optical Window
The placement of Cy7’s fluorescence within the NIR region is of paramount biological significance. Between 650 nm and 900 nm, light absorption by hemoglobin, water, and lipids is minimal, creating a “therapeutic window” where photons can penetrate tissues deeply without significant attenuation. Consequently, Cy7-labelled probes can be visualized through several centimeters of tissue, making them ideal for whole-animal imaging studies, intraoperative guidance, and deep-tumor visualization. Furthermore, the absence of endogenous NIR fluorescence in most biological specimens ensures exceptionally low background signals, dramatically improving the signal-to-noise ratio in imaging experiments.
Find out more about fluorescent peptides here.
Conjugation Chemistry and Methodologies
NHS Ester Chemistry for Amine Labelling
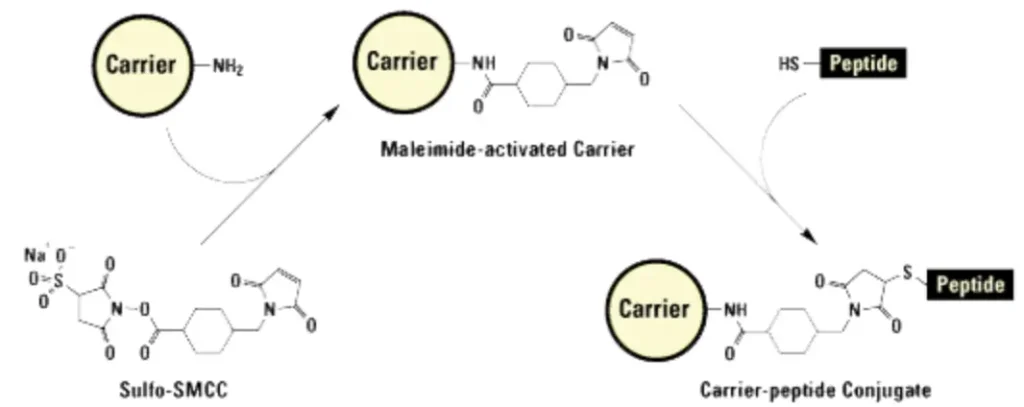
The most prevalent strategy for Cy7 conjugation targets primary amine groups present on lysine residues or protein N-termini. This approach utilizes Cy7-NHS ester derivatives, where the N-hydroxysuccinimide moiety acts as a leaving group upon nucleophilic attack by the amine. The reaction proceeds efficiently under mild, weakly alkaline conditions (pH 7.4–8.5), forming a stable amide bond that covalently links the dye to the biomolecule. Researchers must carefully control the molar ratio of dye to protein, typically ranging from 3:1 to 10:1, to achieve optimal labelling density while avoiding excessive modification that could compromise biological function.
Maleimide Chemistry for Thiol-Specific Conjugation
For applications requiring site-specific labelling, maleimide-functionalized Cy7 offers an elegant solution by targeting the thiol groups of cysteine residues. This Michael addition reaction proceeds rapidly under physiological conditions and provides positional control when cysteines are strategically introduced into peptide sequences. In some cases, mild reduction may be necessary to expose previously oxidized or disulfide-bonded thiols before conjugation.
Reaction Optimization and Purification
Successful Cy7 labelling demands meticulous attention to reaction parameters. Temperature control, typically maintained at 4–25°C, prevents protein denaturation while ensuring adequate reaction kinetics. Light protection throughout the procedure is essential to prevent photodegradation of the dye. Following conjugation, removal of unreacted dye is accomplished through gel filtration chromatography, dialysis, or HPLC purification, yielding high-purity conjugates suitable for sensitive biological applications.
Applications in Biomedical Research
In Vivo Imaging and Biodistribution Studies
Cy7’s deep-tissue imaging capabilities have made it indispensable for tracking biodistribution, tumor targeting, and pharmacokinetics in living animals. Fluorescently labelled peptides and proteins administered to murine models can be non-invasively monitored over time, providing real-time insights into accumulation patterns at target sites. For example, Cy7-conjugated LPETGG peptides have been employed to visualize immune cell interactions in preclinical cancer models, leveraging the dye’s NIR emission to penetrate through tissues and reveal dynamic cellular processes.
Photodynamic and Photothermal Therapy
Beyond imaging, certain Cy7 derivatives function as potent photosensitizers for cancer therapy. Upon NIR light activation, these molecules generate reactive oxygen species (ROS) or heat, inducing apoptosis in targeted tumor cells. Recent innovations have produced asymmetric Cy7 dyes with remarkably high singlet oxygen quantum yields (ΦΔ up to 1.84), enabling effective photodynamic therapy at previously unattainable depths. Importantly, these agents exhibit cancer cell specificity by leveraging microenvironmental features such as elevated viscosity, while demonstrating negligible dark cytotoxicity.
Studying Cell-Cell Interactions with LIPSTIC
In immunology research, Cy7-labelled LPETGG peptides have proven instrumental in the LIPSTIC (Labelling Immune Partnerships by SorTagging Intercellular Contacts) technique. This elegant method uses bacterial sortase A to enzymatically transfer fluorescent dyes from the LPETGG substrate onto interacting cell surfaces, enabling researchers to track dynamic immune partnerships in vivo and in vitro with single-cell resolution. Such applications underscore the versatility of Cy7 beyond simple structural labelling.
Selecting Cy7 for Your Research
Advantages Over Shorter-Wavelength Dyes
When compared to visible-light fluorophores like Cy3 or fluorescein, Cy7 offers distinct advantages for whole-animal studies, deep-tissue imaging, and multiplexing experiments where spectral separation is required. Its NIR emission avoids overlap with common fluorescent proteins and organic dyes, facilitating multicolor panels.
Availability from Commercial Sources
High-quality Cy7 derivatives and pre-labelled peptides are readily available from specialized suppliers. LifeTein, for example, offers Cy7 conjugation on custom peptides such as the LPETGG motif, providing researchers with versatile tools for sortase-mediated labelling and imaging applications. These products undergo rigorous quality control, including HPLC and mass spectrometry validation, ensuring reproducibility in demanding experiments.
Find out more about peptide synthesis here.
Frequently Asked Questions (FAQ)
What are the exact excitation and emission maxima for Cy7?
Cy7 exhibits peak excitation at approximately 749 nm and peak emission at approximately 776 nm, placing it squarely within the near-infrared window optimal for deep-tissue imaging.
How does Cy7 compare to Cy5 or Cy5.5 for in vivo work?
While Cy5 (670 nm emission) and Cy5.5 (701 nm emission) are excellent for many applications, Cy7’s longer wavelength offers superior tissue penetration and lower background due to reduced scattering and absorption by endogenous chromophores. The choice depends on the required depth of imaging and compatibility with available instrumentation.
What conjugation chemistries are available for Cy7?
Cy7 is commonly supplied as an NHS ester for amine coupling or as a maleimide derivative for thiol-specific labelling. The NHS ester is preferred for lysine residues and N-termini, while maleimide enables site-specific conjugation to engineered cysteine residues. Site-specific conjugation via click chemistry is also an option using methods such as Cy7-DBCO and a lys(N3) residue.
Can Cy7 be used for photodynamic therapy?
Yes, certain Cy7 derivatives function as effective photosensitizers, generating reactive oxygen species upon NIR light activation. Recent advances have produced dyes with exceptionally high singlet oxygen quantum yields, making them suitable for photodynamic ablation of tumors.
How stable are Cy7-labelled peptides during storage?
Cy7 conjugates should be protected from light and stored desiccated at -20°C for long-term stability. Reconstituted materials may be stored for up to two weeks at -20°C in aliquots to avoid repeated freeze-thaw cycles.
Are Cy7-labelled peptides available commercially for research?
Yes, specialized providers such as LifeTein offer custom synthesis of Cy7-labelled peptides with high purity (>95%) and rigorous analytical validation. These products are suitable for in vivo imaging, flow cytometry, and advanced techniques like LIPSTIC.
Long, L., Cao, X., Shi, X., Zhang, J., & Shi, C. (2025). Modifications and applications of heptamethine cyanine (Cy7) dyes as near-infrared photosensitizers. Coordination Chemistry Reviews, 541, 216780. https://doi.org/10.1016/j.ccr.2025.216780
Khaikate, O., Muangsopa, P., Piyanuch, P., Khrootkaew, T., Wiriya, N., Chansaenpak, K., Sukwattanasinitt, M., & Kamkaew, A. (2024). Asymmetric heptamethine cyanine dye for viscosity detection and photodynamic therapy. Journal of Photochemistry and Photobiology A: Chemistry, 453, 115659. https://doi.org/10.1016/j.jphotochem.2024.115659