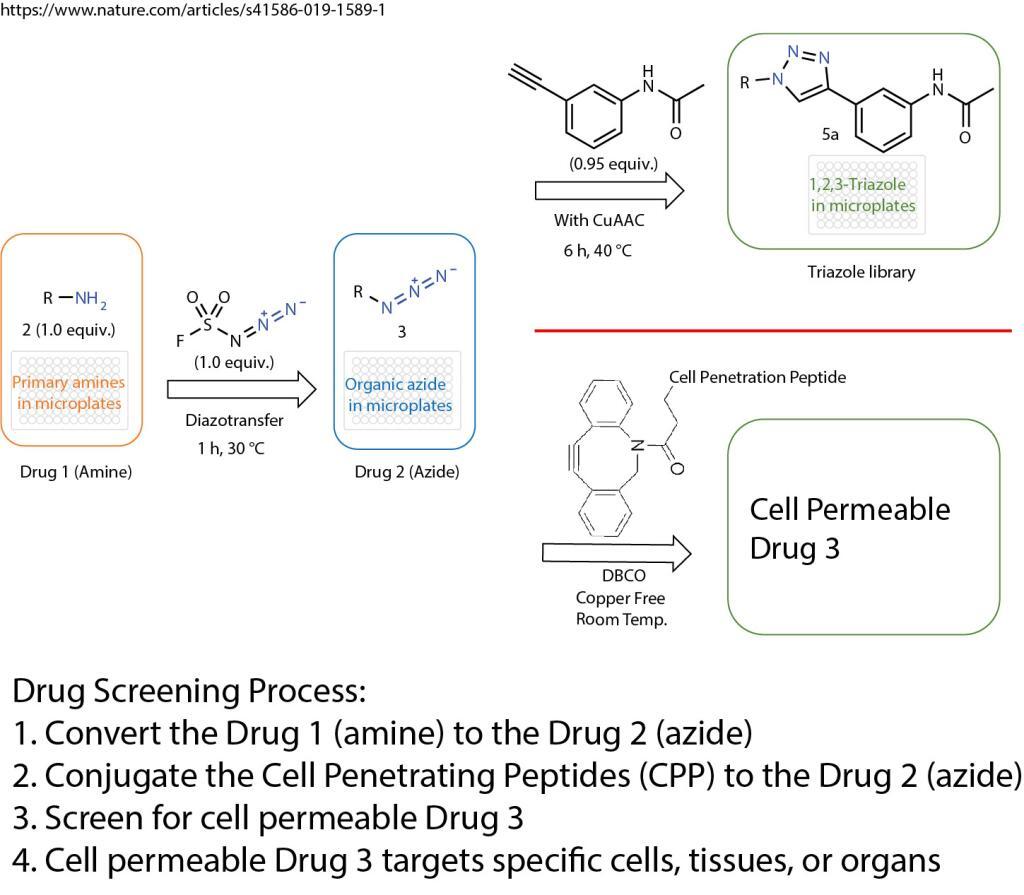
A click chemistry was reported about the formation of azides from primary amines. This powerful tool enables the reaction of just one equivalent of a simple diazotizing species, and fluorosulfuryl azide (FSO2N3), for the preparation of over 1,200 azides on 96-well plates in a safe and practical manner. This method greatly expands the number of accessible azides and 1,2,3-triazoles because the primary amine is one of the most abundant functional groups in small compounds, proteins and antibodies.
Formation of Azides From Primary Amines
The method opens the door for numerous applications in drug screening and discovery. The cell penetration peptides can be easily introduced to conjugate with any azide containing drugs, compounds, antibodies, or proteins.
The cell penetration peptides (CPPs) are capable of delivering biologically active cargo to the cell interior. The desired therapeutic cargo could be attached to a CPP using the copper free click chemistry and then delivered to an intracellular target, thereby overcoming the entry restrictions set by the plasma membrane.
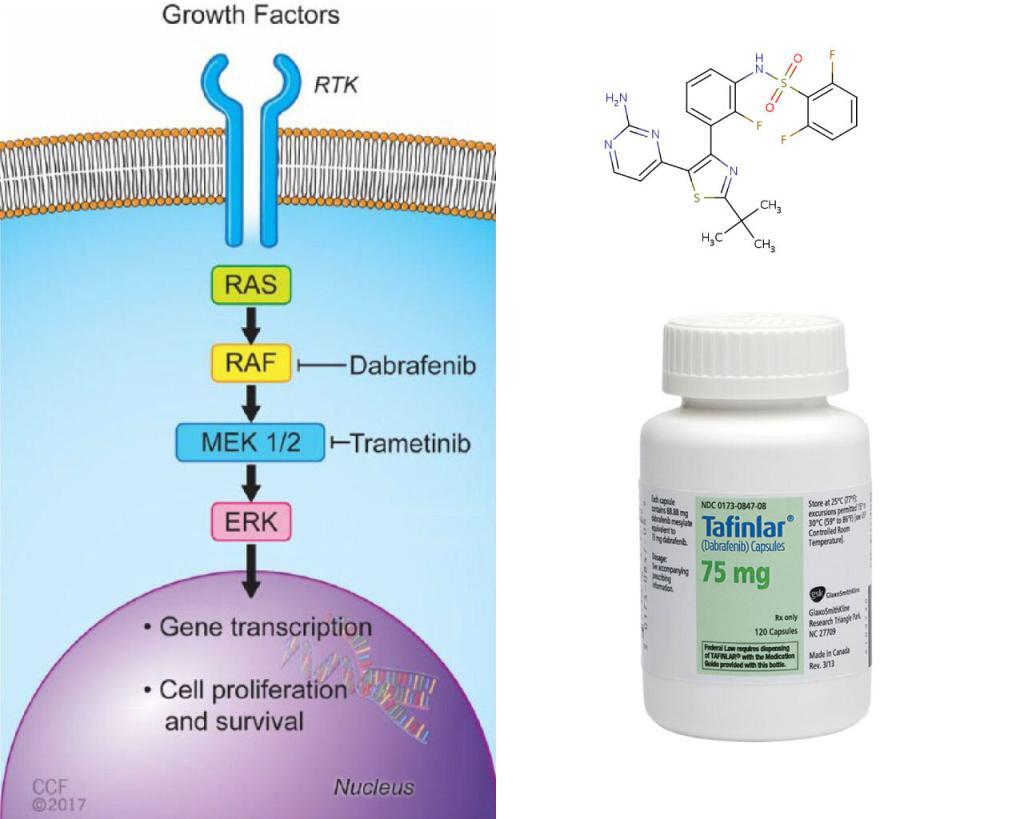
BRAF is an RAF kinase. It is a core component of the RAS/RAF/MEK/ERK signaling cascade, known as mitogen-activated protein kinase (MAPK) pathway. It is one of the major effectors of oncogene RAS, and is often mutated in human cancer cells.
LifeTein’s Braftide & Cancer Therapy
Two FDA approved drugs, Dabrafenib, and vemurafenib, effectively inhibit the most common BRAF variant V600E, a monomeric BRAF. But, the non-V600E BRAF mutations are intrinsically resistant to these drugs. These drugs may also paradoxically stimulate the pathway when the tumor cells contain wild-type BRAF and oncogenic RAS, causing secondary malignancies. The researchers tried to tackle the dimeric BRAF. The dimeric BRAF, such as the wild type and G469A, a most prevalent non-V600E variant in lung cancer cells, hinges on dimer interface (DIF), a 20aa span near the tail end of the alpha-C helix of BRAF. The researchers designed Braftide using computational modeling, aiming to block the dimerization. They tested the functionality in vitro, in HEK263 cells and colon cancer cell lines.
LifeTein synthesized Braftide (TRHVNILLFM), Null-Braftide (THHVNILLFM), Cy3-Braftide (TRHVNILLFM-Cy3), TAT-Braftide (GRKKRRQRRRPQ-PEG-TRHVNILLFM), and TAT (GRKKRRQRRRPQ). We reviewed here some of the assays that helped support Braftide as an allosteric inhibitor of BRAF dimer and down-regulator of MAPK signaling pathway for cancer therapy.
1) Cell-free in vitro assay: dose-response curve. First of all, the researchers show that Braftide has a sub-micromolar IC50 for dimeric BRAF. Full-length dimeric BRAF-WT and BRAF-G469A (from HEK293F cells) were used for dose-response curves, and the BRAF activity was probed by pMEK production.
2) Cell-free in vitro assay: Saturation binding assay. The researchers used Cy3-labeled Braftide (Cy3-Braftide) to characterize (KD) the binding of Braftide with dimeric BRAF-WT using fluorescence quantification.
3) Cell-free in vitro assay: Immunoprecipitation (IP). The purpose of IP was to show Braftide disrupted the BRAF dimerization. Braftide was added to HEK293 cell lysate coexpressing V5- and FLAG-tagged BRAF-WT. FLAG-tagged BRAF was pulled down by FLAG antibody-conjugated resin, which was further probed for V5-tagged BRAF. Braftide indeed reduced homodimer BRAF.
4) Delivery of Braftide into HEK cell for BRAF inhibition. Braftide was tagged with cell-penetrating peptide TAT. TAT-Braftide (and its negative control TAT alone) was used to treat HEK293 cells transiently transfected with BRAF-WT and BRAF-G469A. Four hours of treatment resulted in reductions of BRAF, pMEK, MEK (i.e. the MAPK pathway), which were analyzed with respective antibodies by immunoblotting.
5) Delivery of Braftide into cancer cells for BRAF inhibition and cell proliferation inhibition. Two colon cancer cell lines (KRAS-G13D-colon carcinoma) were treated with cell-penetrating TAT-Braftide and assayed for the inhibition of BRAF activity, down-regulation of MAPK signaling, and cell proliferation. All were shown positive, while the negative control TAT alone were negative.
Researchers at the University of California, San Francisco (UCSF), in collaboration with LifeTein, have made a groundbreaking discovery in the field of pain management. LifeTein’s expertise in peptide synthesis was crucial in developing synthetic scorpion toxin peptides that specifically target the “wasabi receptor,” a key player in the body’s response to certain types of pain.
The wasabi receptor, scientifically known as TRPA1, is an ion channel protein that triggers the familiar sinus-clearing or eye-watering sensation experienced when consuming wasabi or cutting onions. This receptor is also implicated in the perception of chronic pain.
The focus of this research is a peptide derived from scorpion toxin, referred to as WaTx. Remarkably, WaTx, synthesized by LifeTein, can activate the TRPA1 receptor, mimicking the pain response to irritants. Unlike other molecules, WaTx has the unique ability to penetrate cell membranes directly, bypassing the need for channel proteins. This property makes it an invaluable tool for studying chronic pain and inflammation.
In addition to its research applications, WaTx holds promise for the development of new, non-opioid pain therapies. It has been observed to induce pain and pain hypersensitivity without causing neurogenic inflammation, a common side effect of many pain treatments.
Expanding the Horizon: Spider Venom and Chronic Pain
Further expanding on this concept, a study titled “Identification and Characterization of ProTx-III [μ-TRTX-Tp1a], a New Voltage-Gated Sodium Channel Inhibitor from Venom of the Tarantula Thrixopelma pruriens” delves into the potential of spider venoms in pain management. This study, conducted by F. C. Cardoso and colleagues, discovered a novel inhibitor, μ-TRTX-Tp1a (Tp1a), from the venom of the Peruvian green-velvet tarantula. Tp1a selectively inhibits human NaV1.
7 channels, which are key contributors to pain perception.
The study found that Tp1a, both in its recombinant and synthetic forms, preferentially targets NaV1.7 channels, offering a new avenue for analgesic drug development. Unlike many other spider toxins affecting NaV channels, Tp1a does not significantly alter the voltage dependence of activation or inactivation of these channels. This unique feature of Tp1a was demonstrated to be effective in reversing spontaneous pain in animal models.
The structural analysis of Tp1a revealed an inhibitor cystine knot motif, common in spider toxins but with distinct pharmacological properties that could be crucial in developing more selective and potent treatments for chronic pain.
Conclusion
The research at UCSF, along with the findings on spider venom peptides and the significant contributions of LifeTein in peptide synthesis, represents a significant step forward in understanding and potentially treating chronic pain. These discoveries highlight the vast potential of natural toxins in medical research, offering hope for more effective and safer pain management strategies in the future.
Reference:
Lin King, J. V., Emrick, J. J., Kelly, M. J. S., Herzig, V., King, G. F., Medzihradszky, K. F., & Julius, D. (2019). A Cell-Penetrating Scorpion Toxin Enables Mode-Specific Modulation of TRPA1 and Pain. Cell. doi:10.1016/j.cell.2019.07.014
In humans, liver-derived insulin-like growth factor (IGF1) drives postnatal growth. Early childhood infection of E. coli, Campylobacter spp., even asymptomatic, reduces IGF1 level and restricts early-childhood growth. Does the pathogen-induced Toll-like innate immune signaling contribute to growth restriction? To answer the question, the researchers examined a corresponding pathway in fruit flies.
LifeTein’s Peptide: FLAG(GS)HA
In fruit flies, Dilps (Drosophila insulin-like peptides) drive their growth, for example, the growth rate of imaginal discs which give rise to adult structures such as wings. Dilps share homology with insulin and IGF1, and they bind to the insulin receptor. Dilp6 is produced by fat body, an organ for nutrient storage and immune functions.
The researchers found Dilp6 is a selective target of Toll signaling in the fat body, an innate immune response from bacterial infections. They also found that Toll signaling reduces Dilp6 transcripts, and dramatically suppresses circulatory Dilp6 levels, and restricts whole-body growth. Restoring Dilp6, on the other hand, rescues growth and viability in fruit flies even with active Toll signaling.
LifeTein’s peptide FLAG(GS)HA was used as a standard in ELISA to quantify Dilp6 in fruit fly hemolymph samples. Here, Dilp6 was tagged with FLAG and HA because of FLAG- and HA-tagged Dilp6HF allele from CRISPR/CAS9. In this ELISA assay, the plate wells were coated with anti-FLAG antibody, then FLAG(GS)HA or fruit fly hemolymph sample were added to the wells. FLAG(GS)HA and FLAG- and HA-tagged Dilp6 were quantified by anti-HA-Peroxidase 3F10 antibody and subsequent chromogenic reaction. For more details of the method, see the section “Hemolymph Dilp6 measurements by ELISA” in the link.
The smaller ions (F- and Cl-, 50mM NaF and NaCl solutions) tend to stabilize β-sheets
Peptide amphiphiles are composed of hydrophobic alkyl tails and peptide regions designed to self-assemble into cylindrical supramolecular nanofibers in solution. While hydrogen bonds form some β-sheets between short β-strands (2 or 3 residues), others are formed by extended-strands.
Smaller Ions Stabilize β-sheets
The strongly-hydrated ions (F- and Cl-) are more attracted to the positively charged lysine residues on the surface of the peptide nanofiber. When peptide residues form β-sheets, an F- or Cl- ion forms a salt bridge between the side chains of lysine residues from two neighboring peptide amphiphile chains. The salt bridge stabilizes the peptide by bringing the backbones closer, resulting in a transition from random coil to extended β-sheets structures. The smaller ions (F- and Cl-, 50mM NaF and NaCl solutions) tend to stabilize β-sheets slightly better compared to the larger ions (I-, Br-).
So, the self-assembly of peptide amphiphiles into supramolecular nanofibers can be regulated by modifying the salt solution.
Hydrogels with a capacity to absorb and hold water within a porous, swelled structure make it a great candidate as a drug delivery system due to their broad range of physical properties as well as chemical adaptability. For example, a positively charged polypeptide (poly-l-lysine, PLL) coupling to a self-assembling dipeptide (Fmoc-FF) leads to the formation of hydrogels with rheological properties suitable for injection.
Self-Assembling Peptide Hydrogels As a Drug Delivery System
Hydrogenation via self-assembly is a hierarchical process. The hydrogels can be easily prepared via simple mixing and incorporated with various stoichiometric ratios of peptides without any additional synthetic processes. Peptides can form hydrogels by multiple non-covalent interactions. It was found that PTZ-Gly-Phe-Phe-Tyr can form gels at ultra-low concentrations of 0.01 wt.%. The π-π stacking may be another driving forces in the self-assembly to form the final hydrogel structure. The N-cadherin mimic peptide (CLRAHAVDIN) and TGF-β1 mimic peptide (CESPLKRQ) synthesized by LifeTein were used to form an injectable 3D hydrogel. Increased retention of stem cells by using an injectable hydrogel has resulted in successful tissue engineering outcomes.
Another efficient method to facilitate hydrogel formation is based on the electrostatic attraction of oppositely charged peptides. The order of amino acids is of great importance for hydrogenation. Self-assembling beta-hairpin peptides, with high arginine content, exhibited extremely good performance in killing bacteria. The peptide hydrogels also have been demonstrated to be used as wound healing agents and other therapeutic instruments under different conditions. Curcumin could be encapsulated into hairpin hydrogels as an injectable agent for localized delivery.
Design a biotin-labeled peptide library: overlapping or Alanine scan
The peptide library is coated on the avidin-coated 96 well plates.
The Peptide library is used to evaluate the binding of each peptide to different cell lines using CyQUANT Cell Proliferation Assays.
Flow cytometry: Determine the cellular uptake of selected FITC-labeled peptides.
Competitive assay: The cells are incubated with FITC-peptide with or without excess unlabeled peptide (100-fold)
Finetune the peptide sequence: structure analysis, cyclization, and D amino acid modification.
Find the promising receptor-specific cancer cell binding peptide that can be conjugated directly to a chemotherapeutic drug or to nanoparticles for targeted drug delivery to enhance the efficacy of chemotherapy for cancer treatment.
Synthetic peptides have proven an excellent type of molecule for the mimicry of protein sites. The modified peptides increase the proteolytic stability of the molecules, enhancing their utility for biological applications.
Toolbox for Peptide Synthesis: Non-Proteinogenic Amino Acids and Site-Selective Ligation
The long peptides can be synthesized by the ligation method. Amino acid derivatives with modified backbone length and side-chain orientation, such as d-amino acids, N-alkyl glycine monomers, or proteolytically stable amino acid derivatives can be introduced to the peptides.
Protein Secondary Structure Mimics: α-Helix Mimics, β-Sheet Mimics Peptide chains can be organized into secondary structures, such as α-helices and β-sheets. Peptides that mimic α-helices and β-sheets of proteins are attractive targets for drug development and tools to explore protein binding mechanism.
The α-helical conformation of a peptide can be induced by adding covalent links between amino acid side chains at selected positions. These links can be formed by lactam and disulfide bridges, triazole-based linkages, and hydrocarbon staples.
In β-sheets, β-strands are connected via loops or turns. Methods to mimic turn structures include macrocyclization, dipeptide of d-proline and l-proline, or α-aminoisobutyric acid in combination with either a d-α-amino acid or an achiral α-amino acid. An example of stimuli-responsive peptides is the temperature-dependent formation of hydrogels by β-sheet peptides. The β-hairpin mimic undergoes gelation upon heating at 60°C, and is completely reversible while cooling.
Protein Mimics in Biomedical Research
Peptides mimicking the CHR region of gp41 were developed to inhibit the formation of the six-helical bundle. Peptides that mimic these receptors are useful tools to explore the details of virus infection mechanism, as well as to develop new drugs against HIV-1. Peptides that mimic the extracellular domains of seven transmembrane G protein-coupled receptors (GPCRs), which is composed of the N-terminus (NT) and the three extracellular loops (ECLs) were explored. Peptide Ac-RERF-NH2 has a high propensity to adopt an α-turn structure and could be a promising drug candidate against cancer.
The design of peptides as protein mimics has evolved as a promising strategy for the exploration of protein-protein interactions, as they are biocompatible, biodegradable, and functionally selective.
Biochemists are excited by the possibilities presented by peptides and proteins as pharmaceuticals because they so often mimic exactly the behavior of a natural ligand – the substance that interacts with the receptor on an enzyme or cell to cause a biological process.
Peptides are used for: 1. Self-Assembling Peptide Epitopes as Novel Platform for Anticancer Vaccination: OVA 250−264 and HPV16 E7 43−57.
2.Incorporation of DSPE-PEG and cRGD-modified DSPE-PEG molecules improves the biocompatibility and cellular uptake of the nanoprodrug platform: Click chemistry conjugation of peptides to PEGs.
To provide the best experiences, we use technologies like cookies to store and/or access device information. Consenting to these technologies will allow us to process data such as browsing behavior or unique IDs on this site. Not consenting or withdrawing consent, may adversely affect certain features and functions.
Functional
Always active
The technical storage or access is strictly necessary for the legitimate purpose of enabling the use of a specific service explicitly requested by the subscriber or user, or for the sole purpose of carrying out the transmission of a communication over an electronic communications network.
Preferences
The technical storage or access is necessary for the legitimate purpose of storing preferences that are not requested by the subscriber or user.
Statistics
The technical storage or access that is used exclusively for statistical purposes.The technical storage or access that is used exclusively for anonymous statistical purposes. Without a subpoena, voluntary compliance on the part of your Internet Service Provider, or additional records from a third party, information stored or retrieved for this purpose alone cannot usually be used to identify you.
Marketing
The technical storage or access is required to create user profiles to send advertising, or to track the user on a website or across several websites for similar marketing purposes.