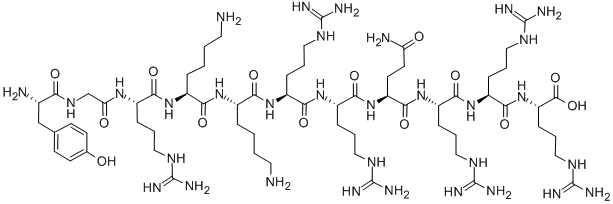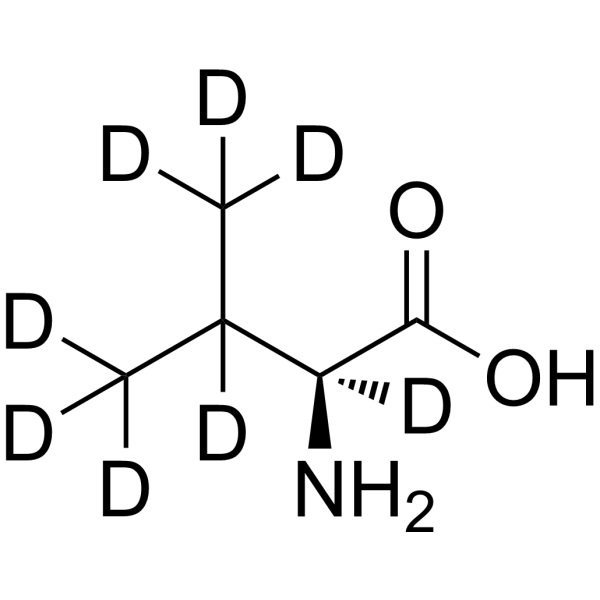
Deuterated peptides represent a significant advancement in the field of drug development and structural characterization. These molecules, where one or more of the hydrogen atoms have been replaced by its heavier stable isotope deuterium, are showing great promise in clinical trials.
Key Takeaways:
- Deuterated peptides have enhanced pharmacokinetic profiles.
- They offer increased bond strength and chemical stability.
- Deuterated peptides have a slower rate of metabolism by liver enzymes.
- They have potential applications in various fields, including drug development and structural characterization.
The Science Behind Deuterated Peptides
What are Deuterated Peptides?
Deuterated peptides are molecules where one or more of the hydrogen atoms have been replaced by its heavier stable isotope deuterium. This isotopic substitution – C-H to C-D – gives rise to the kinetic isotope effect. The C-D bond has a much slower reaction rate than the C-H bond. The kinetic isotope effect is more pronounced because of the percentage difference in mass between deuterium and hydrogen (2 versus 1 in mass).
The Benefits of Deuterium
The extra neutron in deuterium is what adds so many unique benefits to deuterated drug molecules. Deuterated peptides have higher enzymatic resistance, so they have a longer residence time in the body. This reduces the need for frequent dosing. Deuterated versions of drugs also have higher efficacy and lower toxicity due to the delayed formation of toxic metabolites.
Read more on deuteration here.
Applications and Future Prospects of Deuterated Peptides
Therapeutic Applications of Deuterated Peptides
Deuterated peptides have found their way into various therapeutic applications. They are being used in the development of drugs for a wide range of diseases, including cancer, cardiovascular diseases, and neurological disorders. The enhanced pharmacokinetic profiles of deuterated peptides make them ideal candidates for these applications.
Read about LifeTein’s deuterated peptides here.
Deuterated Peptides in Oncology
In the field of oncology, deuterated peptides are being explored for their potential in targeted cancer therapies. The increased stability and slower metabolism of deuterated peptides allow for more precise delivery of therapeutic agents to cancer cells.
Deuterated Peptides in Neurology
Deuterated peptides also show promise in the treatment of neurological disorders. The ability of these peptides to cross the blood-brain barrier makes them potential candidates for the treatment of diseases like Alzheimer’s and Parkinson’s.
The Future of Deuterated Peptides
The future of deuterated peptides looks promising. With ongoing research and development, we can expect to see more deuterated drugs entering clinical trials and eventually making their way to the market. The unique properties of deuterated peptides, combined with the expertise of companies like Lifetein, are paving the way for a new era in drug development.
Frequently Asked Questions
What are Deuterated Peptides?
- Deuterated peptides are molecules where one or more of the hydrogen atoms have been replaced by its heavier stable isotope, deuterium.
Why are Deuterated Peptides Important?
- Deuterated peptides have enhanced pharmacokinetic profiles, increased bond strength, chemical stability, and a slower rate of metabolism by liver enzymes. These properties make them ideal for various therapeutic applications.
What are the Applications of Deuterated Peptides?
- Deuterated peptides are being used to develop drugs for a wide range of diseases, including cancer, cardiovascular diseases, and neurological disorders.