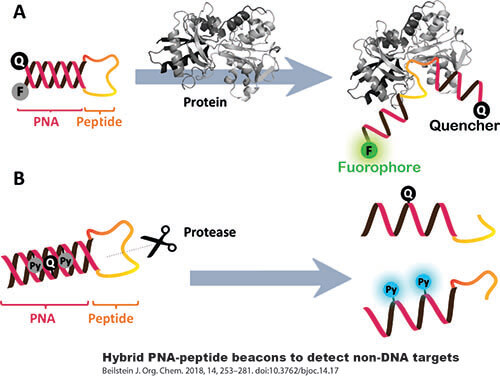
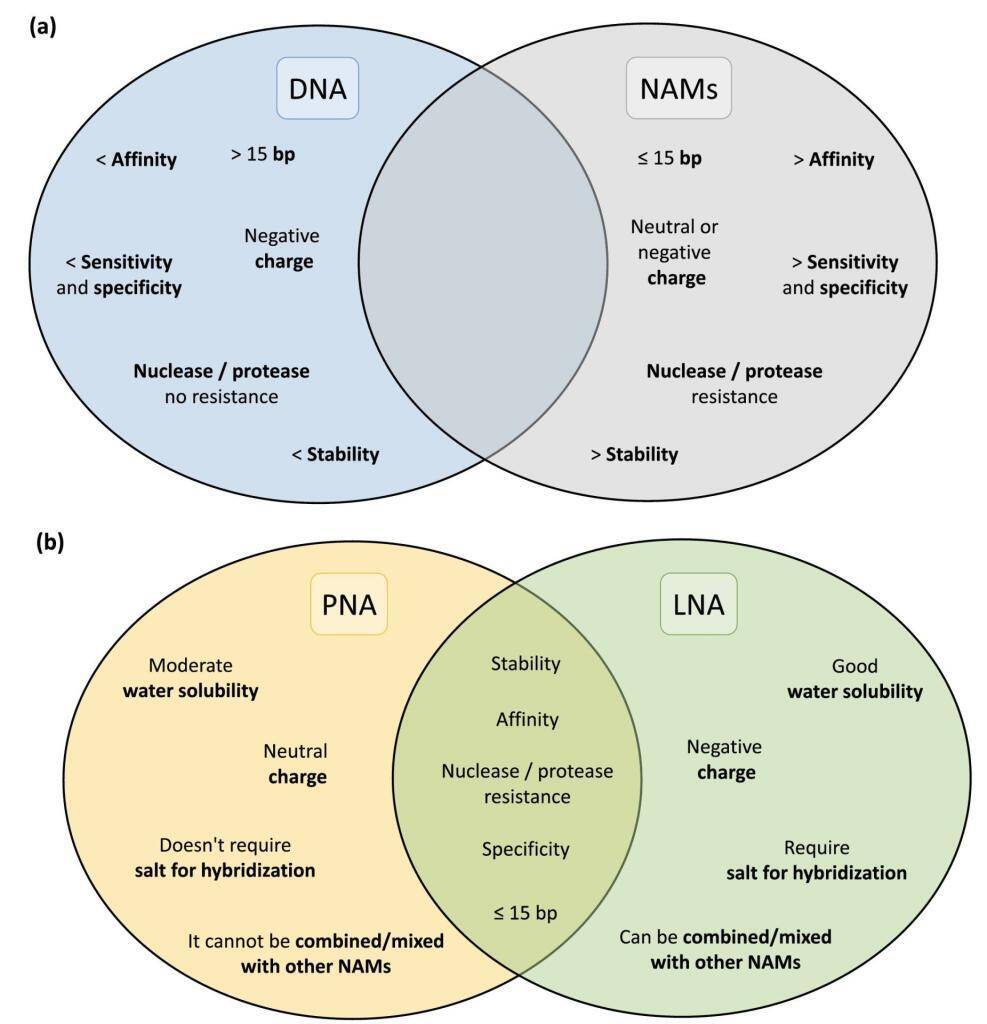
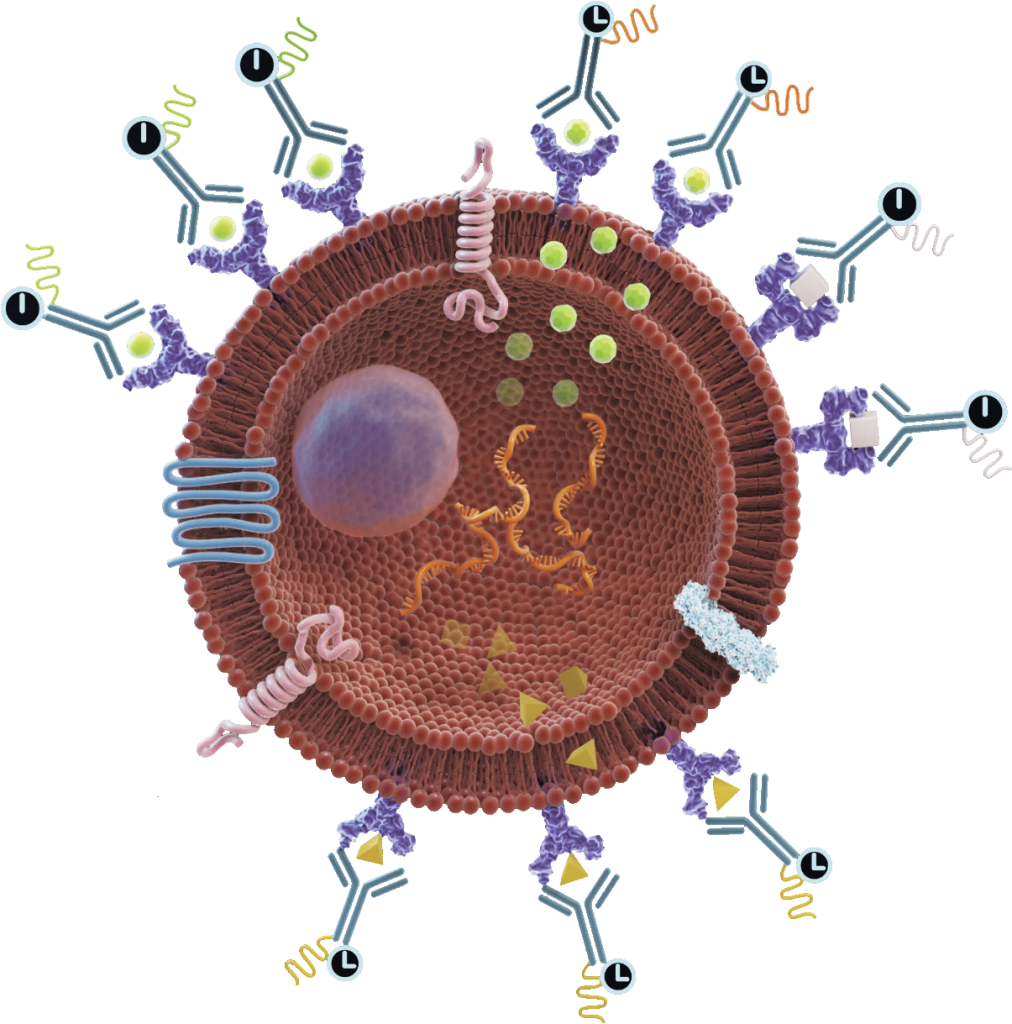
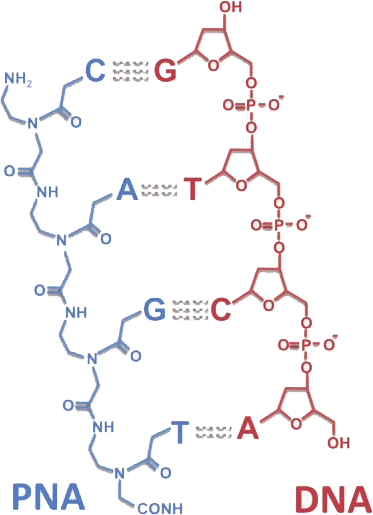
Equipped with exceptional cell permeability and a robust affinity for RNA, LifeTein’s Peptide Nucleic Acid (PNA) synthesis service emerges as the premier platform technology, continuously evolving to meet diverse research needs.
Antisense therapy involves introducing artificial nucleic acids into cells, targeting specific regions of pre-mRNA or mRNA to impede the translation of disease-causing proteins. Over the last five decades, scientists have explored various artificial nucleic acids such as phosphorothioate, 2-alkyloxy RNA, morpholino, locked nucleic acid, and siRNA for therapeutic applications. However, due to their poor cell permeability, these attempts have yielded limited success in developing RNA-modulating drugs with practical utility.
Peptide nucleic acid (PNA) represents a novel class of artificial nucleic acids. Despite its favorable properties resembling natural DNA or RNA oligonucleotides, PNA has remained underutilized in therapeutic applications due to its inadequate cell permeability and physicochemical characteristics.
LifeTein’s PNA synthesis service introduces modifications incorporating cell-penetrating peptides or cationic lipid moieties into the PNA sequence. This strategic attachment significantly enhances cell permeability and augments RNA affinity. Endowed with superior membrane permeability and a potent affinity for RNA, LifeTein’s PNA stands out as the optimal platform for artificial nucleic acid therapeutics.
With its potential to readily traverse the cell membrane, LifeTein’s PNA is ideally suited for modulating the splicing process within the nucleus. Moreover, its tight and selective binding to pre-mRNA effectively prevents spliceosome complex formation, facilitating exon skipping at concentrations orders of magnitude lower than other classes of artificial nucleic acids. While LifeTein PNA can also bind to mRNA and inhibit protein synthesis, its mechanism of action on mRNA requires significantly higher concentrations, limiting its therapeutic viability in this regard.
Representing the third generation of antisense nucleic acids, antisense peptide nucleic acid (asPNA) offers enhanced resistance to natural degradation pathways and exceptional affinity and fidelity towards mRNA targets. Unfortunately, similar to DNA/RNA, asPNA faces challenges in cell internalization due to its large molecular weight and lack of positive charges. LifeTein’s modified peptide nucleic acid addresses these challenges with a rationally designed geometry, customizable functionality, and outstanding biocompatibility, promising significant advancements in biomedical applications.
In summary, LifeTein’s PNA synthesis service offers a multifunctional solution for biomedical research. It leverages tailored modifications to enhance cellular uptake, overcome drug resistance, and achieve effective gene silencing, thereby opening new avenues for therapeutic development in various fields, including tumor therapy.
The landscape of in situ hybridization (ISH), specifically fluorescence in situ hybridization (FISH), has undergone a transformative shift with the introduction of Nucleic Acid Mimics (NAMs). These modified probes, encompassing Peptide Nucleic Acid (PNA), Locked Nucleic Acid (LNA), 2′-O-Methyl-RNA, UNA (unlocked nucleic acid), and Phosphorodiamidate Morpholino Oligomers (PMOs), have emerged as groundbreaking tools, overcoming the limitations associated with traditional DNA and RNA probes.
Peptide Nucleic Acids (PNAs):
Peptide Nucleic Acids (PNAs) stand at the forefront of this molecular revolution, offering a fusion of DNA specificity and peptide versatility. The distinctive PNA backbone, composed of peptide linkages, ensures unparalleled stability and resistance to enzymatic degradation. With superior hybridization properties, PNAs bind to complementary DNA or RNA sequences with exceptional affinity, making them indispensable for applications ranging from targeted gene therapy to diagnostic assays and antisense technologies.
Unlocking the Potential: PNA’s Key Features:
Stability and Resistance: PNAs, characterized by a neutral polyamide backbone, showcase remarkable stability against nucleases and enzymatic degradation. This attribute enhances the half-life of PNA molecules, ensuring their efficacy in diverse experimental conditions.
High-Affinity Binding: The hybridization capabilities of PNAs are unparalleled, facilitating strong, sequence-specific binding to target nucleic acids. PNA’s shorter length allows for enhanced cell penetration and consistent hybridization performance, even under low salt concentrations. Its unique melting temperature response to single nucleotide changes enables precise probe design.
Versatility in Applications: PNAs find applications across a spectrum of research areas, including molecular diagnostics, gene editing, and nanotechnology. Their adaptability for specific sequences and functions makes PNAs an invaluable asset in the molecular biologist’s toolkit.
Locked Nucleic Acid (LNA):
Described in 1997, Locked Nucleic Acid (LNA) boasts a ribose ring locked in a specific conformation, ensuring water solubility and low toxicity. LNA’s design flexibility, including modifications like phosphorothioate, enhances resistance to nucleases without compromising affinity. Combining LNA with 2′-O-Methyl-RNA provides flexibility in melting temperature adjustments for optimized hybridization efficiency.
UNA and Other NAMs:
UNA, an acyclic RNA analog, offers flexibility, though it may impact nucleic acid duplex stability. Modifications like 2′-pyrene and 3′-O-amino-UNA address stability concerns, presenting potential applications in FISH experiments. Additionally, Phosphorodiamidate Morpholino Oligomers (PMOs), characterized by non-ionic properties and resistance to nucleases, have shown success in bacterial and fungal infection detection via FISH.
Challenges and Progress:
Despite the exceptional qualities of NAMs, particularly PNA and LNA, their widespread adoption in FISH for microorganism detection has been slower than anticipated. Challenges include hydrophobicity and water solubility issues for PNA probes, along with a general lack of awareness among laboratories. Nevertheless, studies showcasing PNA’s superior performance over traditional DNA probes underscore the promising potential of NAMs in microbial detection.
Conclusion:
As research in the field progresses, ongoing efforts are addressing the limitations associated with NAMs. Their unique features position them as compelling alternatives for FISH-based microorganism detection, holding the promise of unlocking new frontiers in advanced genetic research.
Live-cell imaging using peptides serves as an invaluable asset in the world of life science research, offering a window into the dynamic lives of cells in conditions closely resembling their natural habitat. By capturing real-time images of living cells, researchers can explore intricate aspects of cell behavior, growth, and movement.
In contrast to the static imaging of fixed cells and tissues, where photobleaching poses a significant challenge, live-cell imaging calls for a different approach. While fixed cell imaging demands high-intensity illumination and prolonged exposure, such conditions prove detrimental to the vitality of live cells. Consequently, live-cell microscopy finds itself in a constant balancing act, seeking to attain image quality while safeguarding cell well-being. This balance often imposes limitations on spatial and temporal resolutions in live-cell experiments.
Live-cell imaging encompasses a wide range of contrast-enhanced techniques in optical microscopy. The majority of investigations rely on various forms of fluorescence microscopy, often in conjunction with transmitted light methodologies. The ongoing evolution of imaging techniques and the development of fluorescent probes ensure that live-cell imaging retains its significance as a critical tool in the field of biology.
The Fundamentals of Fluorescence Microscopy
One of the fundamental considerations is determining the precise amount of excitation light required to obtain a meaningful image. It’s worth noting that high-intensity light, especially in the near-UV range, can harm cells and potentially induce DNA damage. However, in live-cell fluorescence microscopy, the primary source of phototoxicity stems from the photobleaching of fluorophores. A critical protective measure involves deactivating the illuminating light when not in use. Employing shutters to control light exposure proves to be a pivotal element in live-cell imaging.
Additionally, it’s crucial to eliminate undesired wavelengths of light and opt for emission filters that are fine-tuned to maximize the signal. Mitigating photobleaching can be achieved by reducing oxygen levels, and minimizing background fluorescence can be accomplished by excluding phenol red and serum from the culture medium. It’s also essential to prevent any contamination of the illuminating light with even minute traces of ultraviolet or infrared wavelengths. The contained photobleaching chemistry within the β-barrel structure of fluorescent proteins (FPs) or peptides makes them less phototoxic.
The most effective strategy for reducing photobleaching and the associated photodamage is to minimize excitation light exposure by carefully managing exposure time and light intensity while maintaining a satisfactory signal-to-noise ratio tailored to the specific research question.
The Live Cell Imaging Microscope
When selecting an optical microscopy system for live-cell imaging, three key factors come into play: detector sensitivity (signal-to-noise ratio), specimen viability, and the speed required for image acquisition. To optimize the signal-to-noise ratio, it’s crucial to select filters that closely match the spectral profiles of the fluorophores in use. Most epi-illumination microscopes and confocal systems acquire data in four dimensions.
Time-lapse imaging, involving the capture of cellular events over various timeframes, is commonly employed. This technique enables the repetitive imaging of cell cultures at specific time intervals. Wavelengths for excitation and emission filters should be finely tuned to match the fluorophore, thus reducing unnecessary light exposure. To minimize photodamage to the specimen, it’s advisable to use the lowest magnification that suits the specific experiment.
Managing the Microscope Environment
Maintaining a physiological environment is imperative for cultured cells and tissues to behave naturally. Thus, the control of factors such as temperature and tissue culture medium composition plays a critical role in obtaining meaningful data in live-cell imaging experiments. Mammalian cell lines are typically maintained at 37°C. Variations in temperature and vibrations can negatively affect focus stability. To address these issues, options like stage-top incubators and fully enclosed microscopes can be considered. Most tissue culture media are buffered to a physiological pH using sodium bicarbonate and 5% CO2.
Unmasking the Potential of Fluorescence Resonance Energy Transfer (FRET)
Fluorescence resonance energy transfer (FRET) is a remarkable technique that enhances the spatial resolution of fluorescence microscopes to under 10 nanometers. This substantial improvement in resolution makes FRET particularly appealing for studying co-localization events in biological samples, especially within living cells. However, in live-cell studies, the risk of FRET measurements being invalidated by acceptor fluorophore recovery, similar to FRAP experiments, must be carefully considered. Therefore, the use of acceptor photobleaching in live-cell experiments may not always be suitable.
Useful FRET calculator that provides a listing of key FRET pair information: www.fpbase.org/fret/
Furthermore, modern fluorescent dyes, such as the Alexa dye series, offer advantages like enhanced quantum efficiency and brightness, making them indispensable for specific techniques. LifeTein offers an extensive range of fluorescent labeling options, including FITC, FAM, TAMRA, Cyanine Dye Cy3, Cy3.5, Cy5, Cy5.5, Cy7, Cy7.5, EDANS/Dabcyl, MCA, AZDye, BODIPY FL or Alexa Fluor (Alexa488, Alexa532, Alexa546, Alexa594, Alexa633, Alexa647), ATTO Dyes (Atto465, Atto488, Atto495, Atto550, Atto647), and DyLight (DyLight 488, DyLight 550). However, chemical fluorescent dyes often exhibit higher cytotoxicity, possibly due to the cytoplasm’s reduced protection from reactive free radical breakdown products when compared to FPs, where the fluorophore is encapsulated within the FP beta-barrel structure. A range of cell-permeable fluorescent molecules is available for the specific labeling of intracellular organelles.
Navigating Photobleaching
Photobleaching is an inherent aspect of live-cell imaging. Even the most advanced fluorescent molecules convert only a fraction of the absorbed energy into fluorescence. Some of this energy inevitably triggers chemical reactions that lead to fluorophore breakdown. Thus, aside from maintaining a physiological environment and confirming the specific labeling and function of the protein or organelle of interest, the most critical factor for successful live-cell imaging and obtaining meaningful data is the minimization of excitation light. This requires a thorough understanding of the microscope and optimization of components that regulate exposure, wavelength selection, and the collection of emitted photons.
A proper experimental setup is equally vital. Waiting for the visual system to adapt to darkness before attempting to locate a faint sample is advisable, given that modern cameras are often more sensitive than the human eye. However, it’s important to avoid using live camera modes to find your sample. Every photon is precious.
Unfortunately, photobleaching remains an unavoidable challenge, often determining the number of images that can be acquired. There is no simple solution to eliminate photobleaching completely. The specific excitation and emission bands, as well as the intracellular environment, influence the apparent brightness and, consequently, the apparent photobleaching of specific fluorescent proteins.”
Please view this video on how to design cell penetrating peptides. The transcript is listed below.
Transcript
Slide 1:
Thank you for joining me. My topic today is the cell-penetrating peptides. My main focus will be the peptide design, peptide synthesis, and its applications.
Slide 2:
First, let me briefly introduce LifeTein. LifeTein was founded in 2008. We have been in the peptide industry for more than ten years. We specialize in peptide synthesis, chemical synthesis, antibody production, and protein services.
Slide 3:
Our main focus is peptide synthesis service. However, over the years, we have expanded to other protein-related areas like protein, antibody services, and products.
Slide 4:
So let us quickly get into the topic: cell-penetrating peptides. What is a cell-penetrating peptide or cpp? From the definition, CPP is a short peptide. They can be about 4-40 aa. The short peptide can enter the cell membrane. They can deliver bioactive cargoes.
Slide 5:
CPPs can also be used to deliver bioactive cargos like siRNAs, DNA, polypeptides, liposomes, nanoparticles, and others, in cells for therapeutic or experimental purposes.
Slide 6:
There are a few popular models for CPP’s entry. 1. The inverted micelle model. The CPPs are positively charged. They interact with the negatively charged phospholipids in the membrane. 2. Direct entry or direct translocation. For example, sequences with multiple Arginines can cause a short-time membrane cytolysis and enter the cells directly. 3. By the traditional method of endocytosis. I will not talk about the details.
Slide 7:
Here are a few examples of the CPP. The most famous examples are HIV tat sequence. The TAT peptide is arginine-rich and can directly penetrate the plasma membrane and stabilize DNA.
Another example is the arginine-rich peptide R8 or R9. We can add stearic acid to the N-terminus. Stearic acid is a saturated fatty acid with an 18-carbon chain. If you would like to do live cell imaging, we can add fluorescent dye such as Fitc, Alexa fluor, or Cy dye at the N-terminus or C-terminus. I will get to the details.
Slide 8:
CPPs can enter the regular cell membranes. Some other peptides are tissue-targeting peptides. For example, this brain-homing peptide can cross the blood-brain barrier. Other peptides can cross the skin as transdermal peptides, target heart tissues as cardiac targeting peptides, and nuclear localization signal peptides.
Slide 9:
On this slide, I will talk more about the peptide design. There are many ways to make the peptide permeable. In the case of DNA or RNA, you can simply mix the CPPs with oligos. Many transfection reagents are using this mechanism. Simply put, DNA is negatively charged and peptide is positively charged. If you mix them together, they will form small micelles for cell penetration.
However, most of our work is to put the CPPs and your target together by covalent links. For this example, we put eight arginines at the N-terminus of your peptide. A linker called Ahx is added as a spacer. Some users prefer no spacers. It seems that both worked for the purpose. The eight arginines can be put at the C-terminus as well. According to the feedback from our users, most of the N-terminal CPP worked well. A few worked well for the C-terminal conjugation. I guess it depends on the projects.
This example is the Npys linker modification. The cysteine is added to your peptide. We conjugate two sequences together to form a disulfide bond. This is especially useful for the cancer study. Cancer cells have a lower pH of 6.7-7.1. Normal cells have a higher pH of 7.4. Under the acidic environment, the disulfide bond can be cleaved. If your target peptide is a cancer drug candidate, the CPP can introduce the drug cargo to the cancer cell and release the target within the cell. These disulfide-based prodrugs are important for cancer therapy.
If the cysteine is not available for your case, we can add a compound called lysine azide. This method needs click chemistry.
Slide 10:
There are two kinds of click chemistry. The one with copper as the catalyst and the one without. The preferred method is copper-free click chemistry. It is called DBCO and azide reactions. The final conjugate will have a large linker. Many scientists have concerns about the bulky size of the linker. However, some drugs contain bulky linkers without issues or side effects.
Back to Slide 9:
Let us go back to the sequence. The design does not have to be this way. The lysine azide can be any place in the sequence. If you have a head-to-tail cyclic peptide, you can add the lysine azide in the middle. The final product will be like a lollipop, with the CPP as the tail. If the N-terminus is very important to you, you can add the azide at the C-terminus.
Slide 11:
Let us move on to other scenarios. If you would like to track the peptides in live cells, fluorescent dyes can be added. We can do Fitc, Fam, Cy3, cy5, Cy7 and Alexa Fluor. This design will give direct evidence that your target is inside the cells.
There is a different kind of peptide called peptide nucleic acid or PNA. It is DNA or RNA analog. We can synthesize half as peptide and another half as the PNA.
This structure is the one I just mentioned earlier. The cyclic tumor targeting RGD peptide can be linked with an R8 cell-penetrating peptide to form a lollipop-shaped structure.
Slide 12:
So far, we have mentioned different ways to conjugate the cargo with cell-penetrating peptides. If your targets are nanoparticles or gold particles. Our requirement is to have active groups like a thiol group or a free amine on it. We have to have the active groups react to the cell-penetrating peptides. It is the same requirement for small compounds.
Slide 13:
The last concept I would like to introduce is the antibody-drug conjugate. This concept is widely accepted in the antibody drug industry. There are three important components: an antibody, a cleavable linker, and the drug. Once the antibody binds to the target, the drug is released after the hydrolysis by protease.
Slide 14:
The same concept can be used for the peptides. For this concept, we need to screen the best drug candidate for cell entry. The CPP can be tumor-homing peptides, brain-homing peptides, or cardiac targeting peptides I mentioned earlier.
Slide 15:
First, we need to modify the compound. It is better to have a free amine in the compound. Then we can modify the amine group to an azide group. Afterward, we can use the click chemistry for the following conjugation.
Slide 16
LifeTein produced a series of CPPs. They are ready to conjugate your compounds for screening. So far, we have designed and produced more than fifty CPPs.
Slide 17
Step 3 is conjugate peptides with drug candidates.
Slide 18
Once the CPP is conjugated with the drug compound using click chemistry, we can send the final product back to you for further screening. The purpose is to find the best drug delivery system.
Slide 19
To summarize today’s topic, I talked about cell-penetrating peptides with different cargos. As long as you have an active chemical group on the nanoparticles, compounds, or liposomes, we can conjugate the target to any peptide.
Slide 20
That is all for today. Please let me know if you have any questions. Please feel free to contact us by email or phone calls.
In the world of peptide synthesis, a game-changing innovation has emerged – a remarkable cocktail designed to enhance the cleavage and deprotection of methionine-containing peptides. This groundbreaking concoction, known as Reagent H, is set to transform the landscape of solid-phase peptide synthesis, particularly for those using the 9-fluorenylmethoxycarbonyl (Fmoc) methodology.
Unveiling Reagent H: Your Key to Methionine Side-Chain Protection in Methionine-Containing Peptides
Reagent H, comprised of trifluoroacetic acid (81%), phenol (5%), thioanisole (5%), 1,2-ethanedithiol (2.5%), water (3%), dimethylsulphide (2%), and ammonium iodide (1.5% w/w), has been meticulously crafted to minimize the pesky oxidation of methionine side chains during synthesis. Its exceptional performance is exemplified in the synthesis of a model pentadecapeptide from the active site of DsbC, a pivotal player in protein disulfide bond formation.
The Triumph of Reagent H: Methionine Sulphoxide Conquered
When put to the test, Reagent H outshone its competitors, cocktails K, R, and B. The crude peptides obtained from these widely used mixtures contained a staggering 15% to 55% of methionine sulphoxide. However, Reagent H demonstrated its prowess by yielding pristine peptides devoid of methionine sulphoxide. Remarkably, even when 1.5% w/w NH4I was added to cocktails K, R, and B, they couldn’t match the perfection achieved by Reagent H, although their yield of the desired peptide fell short.
Unraveling the Mysteries: A Closer Look at Methionine-Containing Peptides
But how does Reagent H achieve this remarkable feat? We delve into the proposed mechanism behind its in situ oxidation of cysteine, shedding light on its impressive ability to safeguard methionine side chains while delivering high-quality peptides.
In the world of peptide synthesis, Reagent H stands as a beacon of hope for researchers seeking purity, precision, and protection in their work. Its ability to minimize methionine side-chain oxidation is nothing short of revolutionary, promising a brighter and more efficient future for peptide synthesis enthusiasts. Say goodbye to impurities and hello to perfection with Reagent H.
Peptide nucleic acids (PNAs) are synthetic mimics of DNA. The deoxyribose phosphate backbone of PNAs is replaced by a pseudo-peptide polymer. These specific physicochemical properties are exploited to develop a wide range of powerful biomolecular tools, including molecular probes, biosensors, and antigene agents. The PNA molecules can routinely be labeled with biotin, azido, cell penetration peptide fragments, or fluorophores such as FITC, Cy3, Cy5, Cy7, Alexa Dyes, and pyrene.
The uncharged synthetic backbone provides PNA with unique hybridization characteristics. It gives higher stability, or a higher thermal melting temperature (Tm) to the PNA–DNA or PNA–RNA duplexes than the natural homo- or heteroduplexes. In addition, the unnatural backbone of PNAs is not degraded by nucleases or proteases.
It was shown that the binding of PNA to complementary DNA can efficiently block transcriptional elongation and inhibit the binding of transcriptional factors. Thus, the PNAs can be used as antisense or antigene therapeutic agents. PNAs can be used as adapters to link peptides, drugs, or molecular tracers to plasmid vectors. One concept is to form the duplexes of PNAs – cell penetration peptides. The duplexes can penetrate into cells and be used in anticancer applications. The nuclear localization signal (NLS) peptide-PNAs duplexes gave a much higher nuclear localization of a coupled nuclear localization signal than did the free oligonucleotide.
Peptide nucleic acids (PNAs) DNA Complex
The strategy of PNA-directed PCR clamping is used to inhibit the amplification of a specific target. This PNA–DNA complex formed at one of the primer sites effectively blocks the formation of the PCR product. The procedure can be used to detect single base-pair gene variants for mutation screening and gene isolation. The biotinylated short PNA probes can be used as generic capture probes for the purification of nucleic acids via streptavidin beads. Other applications could be solid-phase hybridization, and fluorescence in situ hybridization (PNA-FISH).
PNA-based applications benefit from the unique Physico-chemical properties of PNA molecules, enabling the development of cell penetration peptide-PNA assays in molecular genetics.
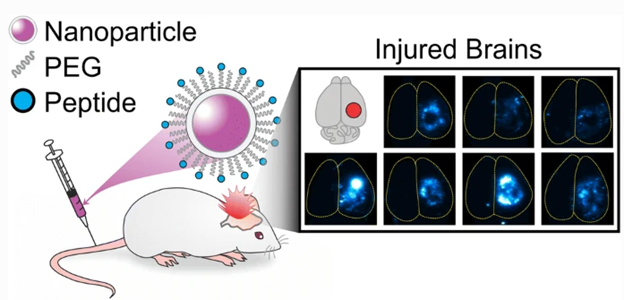
LifeTein’s cysteine-containing peptides helped researchers better understand the effect peptide physicochemical properties have on the pharmacokinetic profiles of the nanoparticles they are attached to. Better understanding and control of these effects is invaluable for future engineering design of many types of therapeutic nanomaterials, including for treatment of traumatic brain injuries (TBI).
Nanoparticles’ Physicochemical Properties Influenced by Peptides
Scientists at the University of California were keen on finding out what exact physicochemical properties in peptides affect the pharmacokinetics of nanoparticles designed for treating TBI. Nanoparticles are a convenient means of therapeutic drug delivery, as they can exhibit different pharmacokinetic profiles from the drug cargo in their core. This experiment analyzed how functionalizing the nanoparticles with PEG and an array of peptides with varying physicochemical properties, provided by LifeTein, contribute to the biodistribution in vivo, using a mouse model of TBI.
Results showed that the biodistribution of the modified nanoparticles varied mainly as a result of the charge of the peptides attached; basic peptides resulted in restricted distributions in the brain via convection-enhanced delivery (CED), as well as elevated off-target organ accumulation resulting in a decrease in brain accumulation when using systemic administration. In comparison, nanoparticles modified with acidic, zwitterionic, or neutral peptides demonstrated less restricted distribution in the brain via CED, and increased accumulation in injured vs. uninjured brain tissue after systemic administration.
This study suggests that the charge of peptides should be greatly taken into account when designing nanoparticles with peptide-modified surfaces. Peptides offer a great way to influence the biological interactions of nanoparticles, and understanding what physicochemical properties contribute to said influence will further advance the use of therapeutic nanoparticles in treatments like TBI.
Reference: Waggoner, L.E., Madias, M.I., Hurtado, A.A. et al. Pharmacokinetic Analysis of Peptide-Modified Nanoparticles with Engineered Physicochemical Properties in a Mouse Model of Traumatic Brain Injury. AAPS J 23, 100 (2021). https://doi.org/10.1208/s12248-021-00626-5
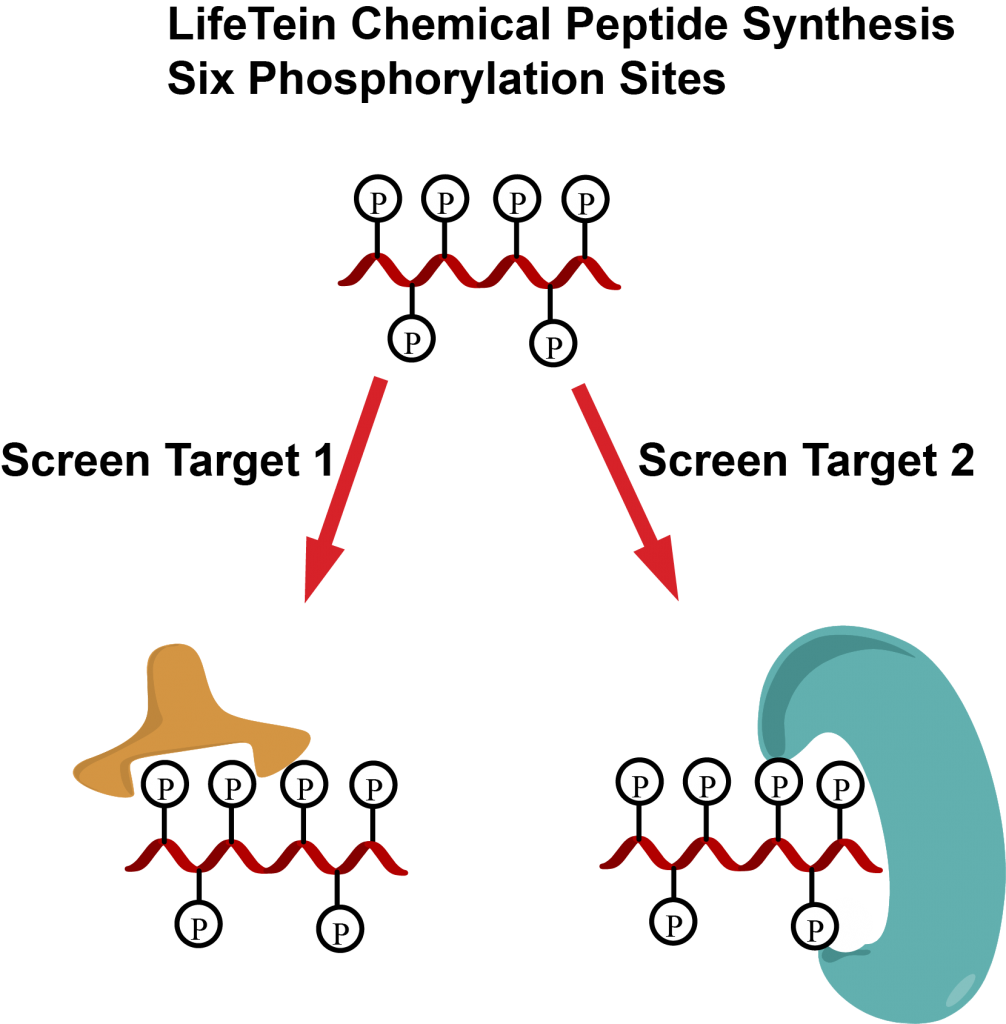
Phosphorylation plays a vital role in regulating cellular processes. Each phosphorylation pattern leads to different interactions and could result in distinct biological outcomes.
Single phosphorylated peptides are frequently used for research. However, multi-site phosphorylations play a more important role in protein-protein interaction, nuclear import and export, and many other functions.
Monophosphorylated peptides are much easier to synthesize than multiphosphorylated peptides using traditional methods. There are many reasons for the difficulties. First, the steric hindrance of the protected phosphorylated amino acids decreases the coupling yield. In addition, the protecting group and cleavage conditions are very harsh for conventional methods.
LifeTein adapted the microwave-assisted heating technology for multiphosphorylated peptide synthesis. Our method increases the coupling efficiency with good yields. In our study, we utilized the optimized conditions for synthesizing peptides containing six phosphorylated amino acids.
Chemical peptide synthesis allows the preparation of multi-phosphorylation peptides with specific required patterns and can be used to elucidate the function of each modification for drug target screening
BRAF is the most frequently mutated kinase in human cancers and is one of the major effectors of oncogenic RAS. So BRAF is a target of interest for anti-cancer drug development and peptide inhibitors.
Two FDA-approved inhibitors, dabrafenib and vemurafenib have been developed as inhibitors for BRAF. These ATP-competitive inhibitors potently inhibit the most common BRAF variant V600E. However, current BRAF inhibitors could induce drug resistance and paradoxical activation. New approaches and drug candidates are needed to disrupt the intact dimer interface of BRAF. The 10-mer peptide inhibitor braftide was designed using a computational approach to block RAF dimerization. It was found that the peptide inhibitor triggers selective protein degradation of BRAF and MEK through proteasome-mediated protein degradation in cells.
The combination of ATP competitive inhibitors and braftide eliminates paradoxical activation. This alternative strategy will improve the efficacy of current ATP-competitive inhibitors. The RAF dimer interface could be a promising therapeutic target.
Braftide is a 10-mer peptide TRHVNILLFM. Braftide disrupts BRAF dimers and inhibits BRAF kinase activity. Braftide causes degradation of BRAF leading to destabilized MAPK complexes. Braftide synergizes with ATP-competitive inhibitors like Dabrafenib to mitigate paradoxical MAPK activation and downregulate MAPK signaling.
In this study, the Braftide, Null-Braftide, TAT-PEGlinker-Braftide, and TAT peptides were purchased from LifeTein with TFA removal.
LifeTein recently announced a significant accomplishment in peptide synthesis: the successful creation of the C subunit of the ATP synthase. This subunit is a highly hydrophobic peptide composed of 75 residues and plays a crucial role in cellular energy processes. Its structure is characterized by a transmembrane α-helical “hairpin” formation, typically assembled into oligomers of 8-16 units, varying with species, within the mitochondrial inner membrane.
The intriguing aspect of the C subunit is its spontaneous folding into a β-sheet conformation. The peptide’s far-UV circular dichroism (CD) spectrum analysis verified this unique structural formation. Intriguingly, the β-sheet conformation of the c subunit can lead to the formation of aggregates. The process of cross-β aggregate formation was carefully observed using the amyloid-binding dye Tiofavin T (TT). A pivotal discovery in this study was the identification of the essential role of Ca2+ in guiding the folding and self-assembly of the c subunits. This process influences the formation of oligomers, as opposed to fibrils.
Amyloidogenic peptides are known for their ability to form ion channels in lipid bilayers, and the human synthetic c subunit synthesized by LifeTein is no exception. This amyloidogenic peptide forms oligomers capable of creating ion-conducting pores in planar lipid bilayers. Interestingly, these oligomers present similarities in ion channel formation with other amyloids and synuclein, albeit with lower conductances compared to the canonical PTP. These misfolded forms of the c subunit, potentially toxic, could play a significant role in cellular pathophysiology, offering insights into various diseases.
This synthesis and study of the ATP synthase’s C subunit mark a notable advancement in peptide research, contributing valuable knowledge to the field of biochemistry and molecular biology.
LifeTein’s work not only showcases the complexity of peptide synthesis but also opens avenues for further exploration in understanding protein structures and their implications in health and disease.
To provide the best experiences, we use technologies like cookies to store and/or access device information. Consenting to these technologies will allow us to process data such as browsing behavior or unique IDs on this site. Not consenting or withdrawing consent, may adversely affect certain features and functions.
Functional
Always active
The technical storage or access is strictly necessary for the legitimate purpose of enabling the use of a specific service explicitly requested by the subscriber or user, or for the sole purpose of carrying out the transmission of a communication over an electronic communications network.
Preferences
The technical storage or access is necessary for the legitimate purpose of storing preferences that are not requested by the subscriber or user.
Statistics
The technical storage or access that is used exclusively for statistical purposes.The technical storage or access that is used exclusively for anonymous statistical purposes. Without a subpoena, voluntary compliance on the part of your Internet Service Provider, or additional records from a third party, information stored or retrieved for this purpose alone cannot usually be used to identify you.
Marketing
The technical storage or access is required to create user profiles to send advertising, or to track the user on a website or across several websites for similar marketing purposes.